

Alberto Marina, investigador de la U739 CIBERER que lidera Vicente Rubio en el Instituto de Biomedicina de Valencia (CSIC), ha coordinado un estudio que describe la relación estructural del colágeno tipo IV con dos enfermedades raras: una de tipo autoinmune, la enfermedad de Goodpasture, y otra predominantemente renal, el síndrome de Alport.
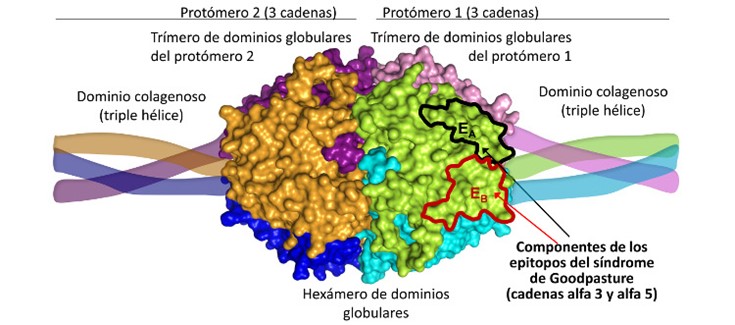
En este trabajo, que acaba de aparecer publicado en la revista más biomédica de la International Union of Crystallography (IUCrJ), los investigadores de la U739 CIBERER, en colaboración con científicos del Centro de Investigación Príncipe Felipe, la Universitat de València y la Universidad de Vanderbilt (Estados Unidos), aportan información estructural clave sobre la enfermedad de Goodpasture, un síndrome raro autoinmune descrito por primera vez en esa última universidad norteamericana en 1919. Este síndrome está causado por la generación de anticuerpos contra los dominios globulares (no colagenosos) de algunas de las seis cadenas del colágeno tipo IV, lo que produce ataque por anticuerpos a las membranas basales de los alveolos pulmonares y del riñón, llevando al fallecimiento o al trasplante renal si no se trata agresivamente.
El colágeno tipo IV forma el andamiaje de las membranas basales sobre las que se asientan las células de los epitelios y endotelios y que rodean incluso las fibras musculares. Controlan gran número de procesos fisiológicos, con impactos también patológicos. Precisamente este trabajo proporciona también una visión de la base estructural del síndrome de Alport, una enfermedad rara renal de origen genético causada por mutaciones en este tipo de colágeno caracterizada por glomerulonefritis y alteraciones auditivas y oculares.
El trabajo desvela la estructura de los componentes de la diana (antígeno) del síndrome de Goodpasture, y, además, revela que las mutaciones de cambio de sentido del síndrome de Alport causan una verdadera enfermedad por mal plegamiento proteico que podría ser tratable mediante chaperonas farmacológicas, moléculas pequeñas que favorecen el buen plegamiento y la estabilidad de las proteínas diana.
El artículo no se restringe únicamente al estudio de situaciones patológicas. También se ocupa de describir cómo se produce el autoensamblado de las redes de colágeno IV desde el punto de vista de la formación de las unidades de seis elementos que son uno de los nodos clave de estas redes. Plantea además la posibilidad de que algunos de los nodos observados en este trabajo, pero no en la naturaleza, estén aún por descubrir en el ser humano. Igualmente sugiere la existencia de maquinarias que ayudarían a la selección de las piezas de colágeno usadas para la formación del andamiaje. Todo ello tiene implicaciones para la función y la patología de las membranas basales.
Aunque la U739 CIBERER ha centrado sus actividades principales en la caracterización de errores metabólicos genéticos raros relacionados con la producción de urea y en la búsqueda de dianas para la generación de nuevos antibióticos que puedan ser de utilidad en enfermedades raras como la fibrosis quística, también se ocupa de otras dianas implicadas en enfermedades raras como es el caso del colágeno tipo IV, de proteínas que producen paraplejia espástica hereditaria, de otras que producen epilepsia dependiente de vitamina B6 o de proteínas involucradas en procesos celulares de señalización.
Sus instrumentos principales de trabajo son las proteínas recombinantes y la determinación de sus estructuras, hasta ahora mediante técnicas de cristalografía de proteínas, a las que ha añadido en la actualidad la criomicroscopía electrónica (objeto del Premio Nobel de Química del 2017 para Jacques Dubochet, Joachim Frank y Richard Henderson) y las técnicas de dispersión de rayos X de bajo ángulo (SAXS).
Artículo de referencia:
“Structures of collagen IV globular domains: insight into associated pathologies, folding and network assembly”. Casino, P., Gozalbo-Rovira, R., Rodríguez-Díaz, J., Banerjee, S., Boutaud, A., Rubio, V., Hudson, B.G., Saus, J., Cervera, J. & Marina, A. (2018). IUCrJ https://doi.org/10.1107/S2052252518012459
(*) Explicación de la imagen:
Las mutaciones missense de la enfermedad de Alport se localizan en zonas cruciales para el plegamiento del dominio globular: se trata de una verdadera enfermedad de plegamiento.




Av. Monforte de Lemos, 3-5. Pabellón 11. Planta 0 28029 Madrid




