

Investigadores de la U713 que lidera José M. Cuezva en el Centro de Biología Molecular 'Severo Ochoa' (CSIC-UAM) han coordinado un estudio colaborativo con otros grupos del CIBERER que ha puesto de manifiesto cómo los pacientes con diferentes Enfermedades Mitocondriales (EMs) tienen la misma alteración metabólica y de daño oxidativo de las proteínas musculares a pesar de la gran heterogeneidad de los defectos genéticos que caracterizan estas enfermedades.
Las EMs son un grupo de enfermedades genéticas raras que surgen como resultado de la disfunción de la mitocondria, la “central energética” y de señalización de la célula. En las mitocondrias tiene lugar la fosforilación oxidativa (OXPHOS), el proceso diseñado para la producción de la energía necesaria para el desarrollo de las funciones celulares. Las EMs son enfermedades progresivas, clínicamente muy heterogéneas, ya que pueden afectar cualquier órgano, especialmente los que se caracterizan por una alta demanda energética como el cerebro, el corazón y el músculo esquelético, dando lugar a un amplio abanico de síntomas que causan discapacidad y, en algunos casos, la muerte.
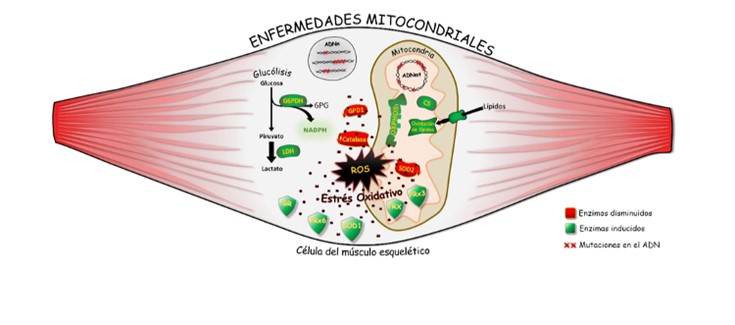
Al igual que las manifestaciones clínicas, el cuadro genético que origina las EMs es muy heterogéneo puesto que las mutaciones que afectan la función mitocondrial tienen lugar en dos sistemas genéticos: el ADN nuclear (ADNn) y el propio ADN mitocondrial (ADNmt). La variabilidad del cuadro clínico y genético que caracteriza este grupo de enfermedades dificulta su diagnóstico, así como la comprensión de los mecanismos patológicos que subyacen a cada tipo de EM.
Este estudio colaborativo del CIBERER en el que han participado la U713, la U723 que lidera Miguel Ángel Martín en el Instituto de Investigación Hospital 12 de Octubre y la U701 que lidera Ramon Martí en el Hospital Universitario Vall d’Hebron ha puesto de manifiesto que, a pesar de la alta variabilidad genética y clínica, los pacientes afectados por diferentes EMs tienen como denominador común el daño causado por el estrés oxidativo a las proteínas musculares, lo que puede ser la causa de las manifestaciones clínicas de estas EMs..
El trabajo, publicado en la revista Free Radical Biology & Medicine, ha sido realizado por Fulvio Santacatterina (U713), Laura Torresano y colaboradores con las muestras de pacientes aportadas por los grupos clínicos que dirigen Miguel Ángel Martín y Elena García-Arumi (U701) y ha sido dirigido por José M. Cuezva. El estudio se ha llevado a cabo mediante la plataforma de microarrays de proteínas en fase reversa (RPPA) PROTEOmAb, localizada en el Centro de Biología Molecular 'Severo Ochoa' y gestionada por el mismo grupo de investigación. Esta plataforma permite un análisis cuantitativo de proteínas del metabolismo, de la respuesta antioxidante y de marcadores de estrés oxidativo, entre otras. Los investigadores han estudiado dos grandes cohortes de pacientes que incluían tres tipos patológicos de EMs. Dos de ellos corresponden a pacientes diagnosticados con oftalmoplejía externa progresiva (PEO), uno debido a una gran delección del ADNmt (PEO-sD) y otro debido a múltiples pequeñas delecciones del ADNmt, supuestamente causada por mutaciones en genes concretos del ADNn (PEO-mD). El tercer grupo corresponde a pacientes diagnosticado con el síndrome de MELAS.
Los resultados han revelado que el músculo esquelético de los pacientes afectados por las tres EMs presenta alteraciones muy similares en la cantidad de algunas proteínas del metabolismo energético y de las enzimas de la respuesta antioxidante, independientemente de los defectos genéticos que causan estas patologías. En concreto, los investigadores han encontrado una fuerte inducción de enzimas del sistema antioxidante como glutatión reductasa (GR), tiorredoxina (TRX), peroxirredoxinas (PRx3 y PRx6) y superóxido dismutasa 1 citoplasmática (SOD1) en una situación paradójica donde las principales enzimas de detoxificación de los radicales libres producidos en la mitocondria y en el citoplasma, como son superoxido dismutasa 2 mitocondrial (SOD2) y catalasa, están fuertemente inhibidas. En esta situación, las proteínas musculares de los pacientes con las tres EMs estudiadas presentan un daño oxidativo mayor que el encontrado en individuos sanos.
En su conjunto, los resultados apuntan a que distintas mutaciones que afectan a las proteínas mitocondriales conducen a la expresión de un fenotipo patológico similar por producir el daño oxidativo de las proteínas del músculo.
Estos descubrimientos contribuyen a esclarecer los mecanismos fisiopatológicos de estas enfermedades y sugieren dos dianas terapéuticas como son SOD2 y catalasa para el tratamiento de las EMs. Los autores creen que estos hallazgos abren una puerta hacia nuevos enfoques terapéuticos que puedan combatir el devastador curso de estas enfermedades.
Fulvio Santacatterina, Laura Torresano, Alfonso Núñez-Salgado, Pau B.Esparza-Molto, Montse Olive, Eduard Gallardo, Elena García-Arumi, Alberto Blazquez, Adrián González-Quintana, Miguel A. Martín, José M. Cuezva. Different mitochondrial genetic defects exhibit the same protein signature of metabolism in skeletal muscle of PEO and MELAS patients: A role for oxidative stress. Free Radical Biology and Medicine, Volume 126, October 2018, Pages 235-248.
https://doi.org/10.1016/j.freeradbiomed.2018.08.020




Av. Monforte de Lemos, 3-5. Pabellón 11. Planta 0 28029 Madrid




